
At-Home DNA Tests
Shop
Genetrack is the trusted name in Saudi Arabia for premium DNA testing services. With our state-of-the-art technology, rigorous quality control, and exceptional service, we ensure the highest standards in laboratory testing.
Product categories
Shop
Bestseller

Find out whether you are the biological father of a child.
- Painless swab sample collection
- All DNA tests are run 2x for 100% accuracy
- AABB, ISO17025 & CLIA accredited lab

Find out whether you’re full siblings, half siblings, or not siblings at all.
- Painless swab sample collection
- All DNA tests are run 2x for 100% accuracy
- AABB, ISO17025 & CLIA accredited lab

Find out whether you are the biological grandparent of a grandchild.
- Painless swab sample collection
- All DNA tests are run 2x for 100% accuracy
- AABB, ISO17025 & CLIA accredited lab

Find out whether you are the biological aunt/uncle of a child.
- Painless swab sample collection
- All DNA tests are run 2x for 100% accuracy
- AABB, ISO17025 & CLIA accredited lab

Find out whether you are biological first cousins.
- Painless swab sample collection
- All DNA tests are run 2x for 100% accuracy
- AABB, ISO17025 & CLIA accredited lab

Conclusively determine twin zygosity (fraternal or identical).
- Painless swab sample collection
- All tests run 2x for accuracy
- Private and confidential

Find out whether you are the biological mother of a child.
- Painless swab sample collection
- All DNA tests are run 2x for 100% accuracy
- AABB, ISO17025 & CLIA accredited lab

Find out whether drinking alcohol is harming your health.
- Understand your genetic risk of alcohol intolerance
- Includes variants of the ADH and ALDH genes

Find out whether your genetic risk of celiac disease.
- Detects 3 HLA celiac risk variants
- Negative results rules out celiac disease for life
- Accurate results even on a gluten-free diet

Determine if caffeine increases your risk of health problems.
- Detects CYP1A2 variants

Determine if you are at risk of lactose intolerance.
- Detects 5 common variants in the MCM6 gene
- Includes lactase intolerance or persistence

Includes Nutrition, Fitness & Weight Loss DNA Tests
- Get 3 tests for the price of 2
- Optimize nutrition, diet, and exercise with DNA

Discover how your DNA influences your fitness journey.
- Identify how DNA influences your exercise motivation
- Discover your body’s pain threshold
- Tailor your workouts to your DNA
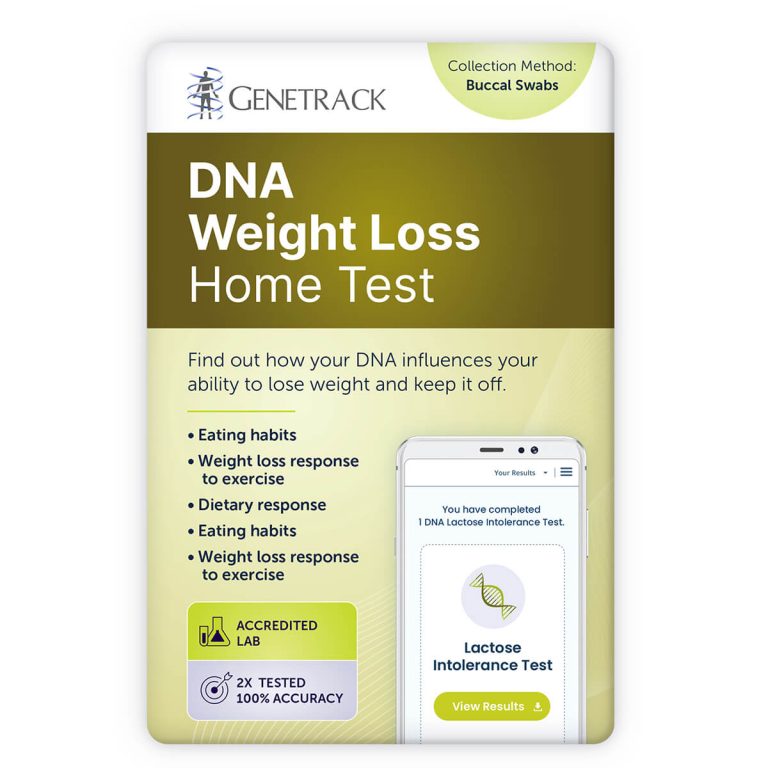
Find out how your DNA influences your ability to lose weight and keep it off.
- Identify genetic factors affecting your weight
- Enhance your weight loss with genetic insights
- Discover your obesity risk based on DNA

Understand your risk of late-onset Alzheimer’s disease.
- Detects variants in the APOE gene
- APOE e4 is a key risk indicator of AD

Determine if you carry the genes linked to iron overload.
- Identifies 3 common mutations in the HFE gene
- 80%+ of patients have two C282Y mutations

Find out if you are genetically at risk for nutritional deficiencies.
- Learn how your body processes vitamins and minerals
- Discover your genetic risk for deficiencies
- Receive DNA-based nutritional recommendations

Are you at increased risk of type 2 diabetes?
- Detects 40+ variants linked to type 2 diabetes risk
- Examines genes affecting glucose, insulin and metabolism

Determine your genetic risk of osteoporosis.
- Identifies variants in 5 key genes linked to bone health
- Includes genes linked to cartilage and bone strength

Understand your genetic risk of cardiovascular disease.
- Detects variants in 35 genes linked to heart health
- Cholesterol, CRP, lipoprotein(a) and triglycerides

Discover your genetic risk of cardiovascular disease.
- Determine if you carry the high-risk ApoE variants
- e2and e4 are linked to increased CVD risk

Determine your genetic risk for dangerous blood clots.
- Find out your genetic risk of thrombosis
- Detects variants in the F5, F2 and MTHFR genes

Optimize your skin health based on your genes.
- Detects DNA changes that influence skin health
- Likelihood of skin disorders & sensitivity to sun
- Uncover effective ways to achieve flawless skin

Determine if your genes increase your risk of narcolepsy.
- Detects the HLA-DQB1*06:02 variant
- 7-25X increased risk with 2 mutations

Find out whether your love of adventure and travelling is in your DNA.
- Detects the “wanderlust” version of the DRD4
- 7R+ variant is linked to seeking new experiences

Find out whether you carry a genetic variant that increases the likelihood of aggressive behavior.
- Detects the “warrior” variant of the MAOA

Determine whether you carry a genetic variant associated with increased sexual promiscuity.
- Determine if you’re genetically inclined to promiscuity
- Analyzes the RS3 repeat region in the DRD4 gene

Find out whether your genes reduce your chance of forming a strong bond with your partner.
- Detects the RS3 334 variant of the AVPR1A
- RS3 334 is linked to difficulty in bonding with partners

Discover your genetic predisposition to infidelity.
- Tests five genetic changes in the AVPR1Agene
- AVPR1A variants are more likely to be unfaithful

Find out whether you carry a genetic variant that disrupts your serotonin levels.
- Tests the 5-HTTLPR variant of the SLC64A